�ֻ�: +86 135 3815 8788 (��������)
Ӣ���绰: +44 790 120 0000 (����/Ӣ������)


Are you ready for Brexit impacts? Do you have a Brexit contingency plan?
You may need either an EU/EC European authorized representative based in EU-27 countries or a UK Authorised Representative (so-called "UK Responsible Person") based in UK, or may need both EU & UK representatives, depending on different brexit scenarios.
Register/Notify your MD-Medical Devices & IVD-In Vitro Diagnostic Medical
Devices with MHRA in UK & other EEA (EU/EFTA) authorities by world-leading
consultancy- Wellkang team based in both UK (England) & EU-27 (Ireland).
Wellkang team can help you under all Brexit scenarios!
Click here to get FREE Guide Now! |
���������κ�һ����ر���Ӷ���ø�Ϊ��ϸ�Ľ��ܣ�
ŷ���漰ҽ����е����ط���
��Ʒָ��(Directives)
ŷ����3�����ҽ����е�IJ�Ʒָ���Щָ����ŷ������EEA��30����Ա��������Ч����3����Ʒָ��Ϊ��
��������
ÿһ��EEA��Ա��������ָ��ר�ŵ����ܻ���CB-Competent Body ��CA-Competent Authority�������侳��ִ��3�����ҽ����е�IJ�Ʒָ�������������ܻ��ؾ��йٷ����ʻ��ٷ����ʡ�
�������(ͨ�����)
ÿһ��EEA��Ա������������ָ��ר�ŵ����ܻ����⣬��ѡ��һ�������ĵ�����(��Ϊ�ǹٷ����ʵ�)������ͨ�������������ͨ�����(NB-Notified Body),������������̶Բ�Ʒָ��ķ����Խ�����������֤����Щ���������ŷ��ίԱ��ͳһ��š��κ�һ������ij�ҹ��������֤���IJ�Ʒ������CE��ǩ���Ա߱���ӡ�ϸù��������(4λ��)���š�
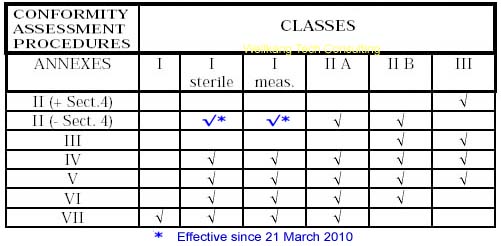
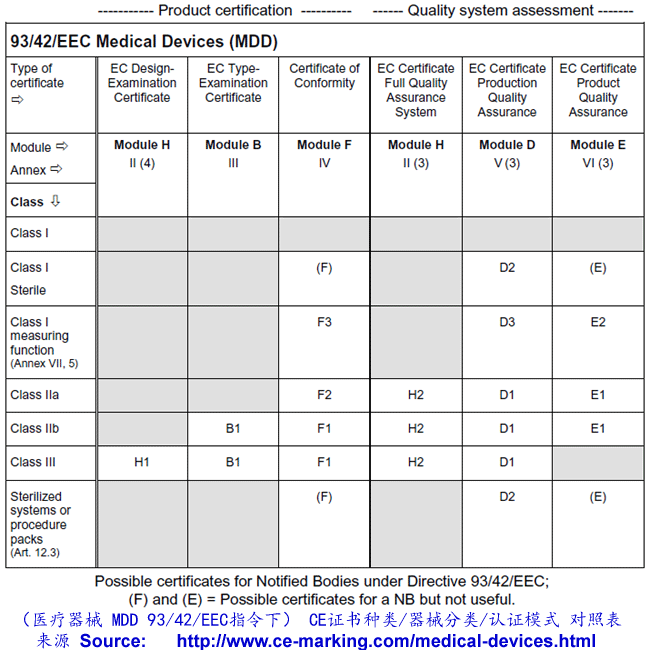
ȡ������ʵ����������һ�������������Ȩ����֤��Χ���ܰ���ij��ָ��������������ģʽ������ֻ�����в��ֻ�һ��ģʽ����ˣ�����һ����������˵����δ��ڶ�Ĺ��������ѡ��һ�����ʵĹ���������������ʺ����Լ�����֤ģʽ���Եø�����Ҫ��
������ص�ָ��
����ļ���ָ��ͨ��Ҳ���漰��ҽ����е:

�ҵIJ�Ʒ����ҽ����е��?
(��ͨ)ҽ����еMDD
����ġ�ҽ����е�������ĵķ�Χ��ͨ�������еġ�ҽ����е����Ķࡣ
�������ҽ����е����ָ�κ���������е�����ߡ����ϻ�������Ʒ(���������������)��
�����ǵ���ʹ�û�������ʹ�ã���������������������Ϊʹ�ö�������;Ϊ����֮һ��
- ��������ϡ�Ԥ�������ӡ����ƻ���
- ���˻���ϵ���ϡ����ӡ����ơ����������
- ����ѧ������ѧ���̵��о����滻��Ľ���
- ����Ŀ��ơ�
����������Ҫ������ҩ��ѧ������ѧ���л���õȷ�ʽ���ﵽ��Ԥ����Ŀ�ģ�
����Щ��ʽ�����������á�
�������ҽ����е (IVD)
���������ҽ����е����ָ������Ԥ�����������������ȡ�õ���Ʒ������ѪҺ����֯����ģ����۵���ʹ�û������ʹ�õ��κ�ҽ����е�������Լ����Լ���Ʒ��У���ϡ����Ʋ��ϡ������ߡ��DZ���װ�á��豸��ϵͳ����Ψһ����ҪĿ�����ṩ������Ϣ��
- �й�����ѧ����ѧ״̬����
- �й��������쳣����
- ����ȷ����ȫ���Լ�����ܽ��������ߵ������ԣ���
- ���ڼ�����ƴ�ʩ��
��Ʒ��������Ϊ���������ҽ����е������Ʒ��������ָ�������ǻ�������͵ģ�����������ȷ�涨��Ҫ����ʢװ�ͱ���������õ���Ʒ��������ϼ�����е��
��ͨʵ�����õIJ�Ʒ�������������ҽ����е�����Ǵ����Ʒ��������������������ȷ�涨����������ϼ���õģ�
������ֲ��ҽ����е (AIMD)
�κξ���������ҽ�Ʒ�����ȫ����ֲ�����壬����벢������������Ȼ����֮����ʽҽ����е��
Ϊʲô���뽫ҽ����е, �������ҽ����е(������������Լ�)��Ϣע��/ͨ��ŷ�����ܻ���?
������ŷ�˵�ҽ����еָ��(MDD 93/42/eec) ���������ҽ����еָ��(IVD 98/79/ec)��Ҫ��ҽ����е�����̻���ŷ����Ȩ����������ע���Ӫҵ�ص����ڵ�ŷ������EEA��Ա�������ܻ���(CA-Competent Authority)��ע�Ტ�ṩijЩ���Ϻ���Ϣ����ЩҪ����ת��Ϊ����ŷ������EEA��Ա���ı������ɡ�
����EEA��Ա�������ܻ���CA���յ�ҽ����е�����̻���ŷ����Ȩ������ע�����룬�������ʵ����ݴ�����Ӧ����֪ŷ��ίԱ�ᡣ��Ҫ�������£�����EEA��Ա�������ܻ���CAӦ����֪����EEA��Ա�������ܻ��� (CAs-Competent Authorities)��
��ע��
EEA��Ա���ڵ������̿����Լ�ֱ�������ܵ��ֽ���ע���ͨ�档��EEA��(�����й���)�������ڼ���CE��־ʱ����Ӧ��ί����ŷ�˴�����ŷ�˴������ڹ������ܵ��ֽ������̼���Ʒ��Ϣ���ע��(Registration)��ȡ��ע��š�
ŷ�˵����ܻ�����ȡע�����?
ŷ������EEA��Ա�������ܻ���CAͨ����ȡע��ѣ���ͬ���ҵ����ܻ����շѷ�ʽ�ͷ���Ҳ��ͬ�����磺
- Ӣ��
���ܻ��ذ������շѡ� ÿ����ȡ70Ӣ�� (Լ��1050�����RMB)�����۲�Ʒ������һ���������һ�ν��շ�70Ӣ��������������ν��շ�140Ӣ����
- ������
ע��һ����Ʒ�����Ϊ455.12Euro (Լ��4550�����RMB)��
- ���
���ܻ��ذ������̼���Ʒ�ͺ�������ȡ��ѡ� ÿһ��������ÿ�������2150���� (Լ��2365�����RMB)������
1��10����Ʒ��ע���Ϊ1000���� (Լ��1100�����RMB)������10����Ʒ����Ʒ�ͺ���������ע��ѡ�
һ��֮�ڲ��۸���/�����Ϣ���ٴΣ�ֻҪ�����Ӳ�Ʒ�ͺ����������������շѡ�
ʲôʱ�����ע��/ͨ�����ܻ���?
- ����(��ͨ)ҽ����еMDD:
���е�һ��(Class I ���������ͼ�����) ҽ����е�����̻���ŷ����Ȩ������������һ�μ���CE��־ʱ����Ӧ���������ŷ�˴������ڹ������ܵ��ֽ������̼���Ʒ��Ϣ���ע�Ტȡ��ע��š����������������IJ�Ʒ�ͺż����ڽ����ڼ���CE��־����Ҳ����һ����ǰ�����ܵ���ע�Ტȡ��ע��š�
����ʽ (custom-made) ҽ����е��ϵͳ���װ�������̣��Լ����������˾�������������״�����������ҽ����еָ��(MDD 93/42/eec)Ҫ��ʱ�����ܵ���ע�Ტȡ��ע��š�
��ע��
�й���һ��(Class I ���������ͼ�����) ҽ����е�����̳��ڼ���CE��־��MDDǰ��Ҫ�����ŷ�˾���ע�ᣬ������뺣��ʱ������������ɾ��ף���ɲ���Ҫ�ľ�����ʧ��
��EEA��Ա��������δ��ע���һ��MDD��Υ������Ϊ������δ��ŷ��ע�������CE��־��һ��MDD��ŷ�˾���ĵ�����Ҳ��Υ������Ϊ��
-
�����������ҽ����еIVDD:
���е��������ҽ����еIVDDs (���� List A, List B, Self-Testing, General/Other) �����̻���ŷ����Ȩ������������һ�μ���CE��־ʱ����Ӧ���������ŷ�˴������ڹ������ܵ��ֽ������̼���Ʒ��Ϣ���ע�Ტȡ��ע��š����������������IJ�Ʒ�ͺż����ڽ����ڼ���CE��־����Ҳ����һ����ǰ�����ܵ���ע�Ტȡ��ע��š�
��ע�� �й���IVDD�������ڼ���CE��־ʱ����Ӧ��ί����ŷ�˴�����ŷ�˴������ڹ������ܻ��ؽ������̼���Ʒ��Ϣ���ע��(Registration)��ȡ��ע���, Ȼ��ʱ���Ѿ�ע�����Ϣ(����CA���Ƽ�ע���)ͨ��(Notify)��������(��Ʒ����������)EEA��Ա�������ܻ��ء�IVDD�����̳��ڼ���CE��־��IVDDǰ��Ҫ�����ŷ�˾���ע�ᣬ������뺣��ʱ������������ɾ��ף���ɲ���Ҫ�ľ�����ʧ��
��EEA��Ա��������δ��ע���IVDD��Υ������Ϊ������δ��ŷ��ע�������CE��־��IVDD��ŷ�˾���ĵ�����Ҳ��Υ������Ϊ��
��ͨ�����ܻ��ص���Ϣ����ˣ���ô��?
���ۺ�ʱ���Ѿ���ŷ������EEA��Ա�������ܻ���ע������Ϻ���Ϣ(���磺���ƣ���ַ����ϵ�绰/���棬��Ʒ��Ϣ����Ʒ����Ͷ���г����ȵ�)һ�б����ҽ����еMDD ���������ҽ����еIVDD �����̻���ŷ����Ȩ��������������֪ͨ������ע������ܻ��أ����ṩ���µ���Ϣ��������صķ��á�
����ŷ��ע���������������Ʊ��ˣ�Ӧ����ô�죿
- ԭ�еļ����ļ�������������ȫ��Ҫ������
ͨ���������ļ��������εظ���(update)����(revise.) ��ÿ�θ��»��ĺ�İ汾(Version)����ע���������ļ����»��ĺ������µİ汾Ϊ��
- ���������Ƹı�����м���CE��־��ҽ����е�İ�װ�ͱ�ǩ�ϵ�����������Ҳ��������������ơ�
- ����������ɹ������ǩ��CE֤��Ļ���CE֤��Ҳ�������µ���������������ǩ����
- ŷ�˷���Ҫ��ŷ����Ȩ�������������ļ����ļ����������°汾����ˣ������̱��뼰ʱ�����°汾�ļ����ļ�����ŷ����Ȩ��������
- ��������̼������CE��־��ҽ����е�Ѿ���ŷ��ע�ᣬע��֤��Ҳ������������ʹ�������Ƶ���������������������ͬһ������(Legal Entity)�������̵�ע�����Ȼ����Ϊԭ���IJ��䡣���ʹ�������Ƶ��������������������ڲ�ͬ�ķ��ˣ���������ŷ�˵�ע���Ҳ���ı䡣
������������̼������CE��־��ҽ����е��ŷ��ע�������⣬��ο�ϵ������ҳ��
http://www.ce-marking.com/authorised-representative
�������ҽ����е(������������Լ�)IVD��ŷ��ע����ͨ��ʱӦ��ע���������
- ���ѡ������ʵ���һ��ע��(First Registration)�Ĺ����Լ����ʵ����ܻ���(Competent Authority):
Ϊ�˱����ٺ�������ŷ��IVD������ͨ��ֻ��ѡ����һ��EEA��Ա����һ�����ܻ���(CA)���е�һע��(registration of placing on the EEA market for the first time)���ɸ����ܻ���CA�������ͨ�����������̼���Ʒ��Ϣ������ŷ�˵����ݿ⣬�����������һ�����õ�ע���(����������FDA�Ĺ��������̵�ע���)��Ȼ�������̲ſ��Խ��Ѿ�ע�������Ϣ������CA����(Competent Authority Code), ����(Competent Authority Name), ע�����ڼ�ע���ͨ��(Notify)��������(��Ʒ����������)EEA��Ա�������ܻ��ء�
һ��ע����������ҽ����е�����̽�����е����ݸ��¼���EEA�г������������뾭�����е�һע������ܻ���CA�����С���ͬ�ij�Ա����ע�����ܻ���CA�ľ��飬������ע����ã���Ҫ���ϣ�����Ҫ��Ҳ���ܴ���ˣ�ѡ��һ�����ʵĵ�һע����Լ����ʵ����ܻ����dz���Ҫ��
- ��(��ŷ����Ȩ������)�����̴����һ��ע��ĺ���
��ŷ�˵�IVDע������ݿ�(European Databank)���6240��Ŀ ���ݽ�ע�����������״���� (status of the organization making this registration application) ���ѡ��ֻ����������֮һ:
- (EEA���ڵ�) ������ Manufacturer (art. 10(1)); ����
- (EEA���������̵�) ŷ����Ȩ���� Authorized Representative (art. 10(3))��
��ˣ�(��ŷ����Ȩ������)��������û���ʸ������һ��IVDע���! ijЩ��������ķ�רҵ�Ľ�������ʱ���Լ�(Distributor) �����ѡΪAuthorized Representative (art. 10(3)), �Ӷ������˵�һ��IVDע�ᡣ�ⲻ�������������Υ��IVDDָ�����ʵ�����һ����������̺ý�����һϵ�еķ���������
- ��ͬ����ע���շѲ�ͬ��
��ͬ�ij�Ա����ע�����ܻ���CA��ע��������ܴ���Ӣ���շ�70Ӣ��(Լ��1050�����RMB)���������3150���� (Լ��3465�����RMB)����������ע��һ����Ʒ�����Ϊ455.12ŷԪ (Լ��4550�����RMB)��
- ��ͬ��������Ҫ��ͬ��
- ע��������ϵ�����
Ϊ�˾����ܵؽ���ʹ���ߵķ��գ�ŷ�˲���Ҫ��ҽ�ƻ�е�ļ����ļ�������ŷ�˵Ĺٷ�����д(ע�����İ�ļ����ļ���Ч), ����ͨ����˵���飬��ǩ�ȱ�������Ͷ���г����صĹٷ����Եİ�(��)��������������ע��ʱҪ�ṩ���صĹٷ����Եİ汾��˵����ͱ�ǩ�ȡ�
����EEAĿǰ��25�ֹٷ�������
��ˣ�ѡ���ú�������дҽ�ƻ�е�ļ����ļ����Լ���һ��������Ϊ��һע������Ե÷dz���Ҫ�����磺���ѡ��Ӣ����Ϊ��һע�����ע���������в���ֻҪӢ�T�ɣ�
- ���ܻ��ط�֤�õ�����
ͨ������һע������ܻ���CAǩ����ע��֤��ȷ�Ϻ���ʹ��ע����Ĺٷ����ԡ���Ȼ��ijЩ��Ӣ����ҵĸ������ܻ���ע�����ݲ�����Ӣ�ļ�ע��������������磺��Ʒ���ƣ��������ְ���շѱ��ȣ���Ȼ��ע����Ĺٷ�������д����ؽ���������̺��������ҵľ�������������Ҫ���鷳������
- �����ܻ��������ź����õ�����
��һע������ܻ���CA������ע����Ϲ����У��Լ����ļල������(���磺������)���侳�ڵ�ŷ����Ȩ����֮������������ź�������ʹ��ע����Ĺٷ����ԣ����磬�����ú�����¹��õ���������������������������ȵȡ�������й���������û�п��ܾ�ͨ��Щ���ԣ�������ֻ�ܽ����ڷ�ҽ����еרҵ��(��Ӣ��)���빫˾����ʱ�������ŷ����Ȩ����Ҳ��רҵ�������̽����ܳ��ܼ���ķ��գ�
- ע��ʱ�漰�ķ����������õ�����
ŷ�˵IJ�Ʒָ��ͨ����ͨ��ת����EEA��Ա���ı������ɶ���Ч����Ա���ı�������ֻ����ٷ����Եİ汾���з���Ч�á������EEA��Ա�����漰ҽ����е�ı���������һ�����ο��õ��������ṩ��Ӣ�İ汾��û�У���ˣ�����й���������ѡ����һ����Ӣ����ҵ�ŷ����Ȩ������һ����Ӣ�������Ϊ��һע������ͱ��뾡��Ƹ�뾫ͨŷ����Ȩ�������ڹ����Ժͷ��ɵ���ʦΪ���ɹ��ʣ��Ա���������ŷ����Ȩ�������������ʱ������Ͷ��ŷ���г��IJ�Ʒ����������ʱ����ʱ��ȡ�ʵ����ж���
����й���������ѡ����Ӣ�����ڵ�ŷ����Ȩ����(���磺Ӣ��ΰ����˾)��Ӣ����Ϊ��һע����������������Ҫ�Ļ��������ҵ��ܶྫͨӢ����������������Ӣ����ͥ��ͥ�������ʦ��
- ��ͬ����Ҫ��ע�����Ҳ�в�����
��ͬ��EEA��Ա����ע�����ܻ��ض�ע����ϵ�Ҫ��Ҳ����ȫ��ͬ��
- ���磬�еĹ��ҵ����ܻ���Ҫ��ŷ���������ڸ�ŷ����Ȩ��������Ȩ���ϵ�ǩ�������ǩ����֤��(����֤����)�ϵ�ǩ����ͬ��ͬʱ�����ṩǩ����֤��(����֤����)�ĸ�ӡ��������������֤�������ܣ������й��������̱����ṩǩ���˻��յĸ�ӡ����
- Ҳ�еĹ��ҵ����ܻ���Ҫ�����ɹ������(Notified Body)ǩ��CE֤���ҽ����е��ע��ʱ������Ҫ�ṩ�������汾��˵����ͱ�ǩ�������������汾��˵����ͱ�ǩ���뾭���������������ȷ�ϣ����ṩ����ȷ�Ϻ���
- ��Ӣ�����
����й���������ѡ����Ӣ�����ڵ�ŷ����Ȩ����(���磺Ӣ��ΰ����˾)��Ӣ����Ϊ��һע��������������շѱ��ˣ����Ҳ���Ҫ�й����������ṩǩ���˻��ո�ӡ����Ӣ��汾��˵����ͱ�ǩ���ɣ�˵����ͱ�ǩ����Ҫ�����������������ȷ�ϡ�
ҽ����е��Ʒ���ڵ�ŷ����Ҫע����?
ŷ�˵ķ���Ҫ��ŷ�˵�ҽ����е�������ڼ���CE��־, ӡˢ��ǩ��˵����ʱ������ӡ�������̺������̵�ŷ�˴��������ƺ͵�ַ�������������ҽ����еIVDD(�����Լ�)�Լ�����I��ҽ����еMDD, �ڼ���CE��־ʱ���ͱ���ί��ŷ�˴����������̼���Ʒ��Ϣ��ŷ�˾���ע�ᣬ����ŷ�����ݿ⡣����ǰ��Ҫ�����ŷ�˾���ע�ᣬ�������ŷ�˺���ʱ�����⡣
�ҹ�˾Ҫ�������Ʒ��CE��֤, ������������Ҫд��Ʒ�ͺ�, �ܶ������е��û���ͺ�, �Dz��ǿ��Բ�д��?
�����Բ�д!
Ϊ�˱�֤��Ʒ�Ŀɸ�����(traceability), ���м���CE��ҽ����е��������ȷ�����ͺ�(������)����������LOT�� ���⣬ŷ������������Υ���Ʒ�ķ����ġ������š� ���ٻ��š� ��������ض������Ʒ�ͺŶ��ԣ�������������еIJ�Ʒ����ʱ����������̻���ŷ�˴������ṩ�㹻��֤�ݣ� ŷ������������Υ���Ʒ�����ġ������š� ���ٻ��š� �������ڽ�������ض������Ʒ�ͺŵ��ض���������LOT���ԣ�������������и��ͺŵIJ�Ʒ��
���������� (DoC- Declaration of Conformity) Ϊ���Т���ҽ����е��Ʒ����CE����Ҫ�������ݣ���ˣ�DoC�в���ʡ�����Ʒ�ͺš�
ΪʲôI��ķ���Ҫŷ�˴�����ȥע�ᣬ��II, III��ľͲ�Ҫ�أ�
ԭ����Ҫ�ж�:
(1)
II, III��IJ�Ʒ�Ѿ��й��������֤. ��I��Ϊ��������, �����������Ҫע�����.
(2)
II, III��IJ�Ʒ��ǩ���Ѿ��й����������, Υ�����¹�ʱ������������ֱ���ҵ������������/����, ����ϵ�����̡�I���Ʒֻ��ͨ��ע���, ��������Ϣ���ܽ���ŷ�����ݿ�, Υ�����¹�ʱ���������ſ��Լ�ʱ��ϵ�������̡�
˭Ӧ�������ܻ�������/ע��?
ŷ������EEA�����ܻ���ͨ����ҽ����е��ע��/ͨ����ȡһ�����á�
- ����һ��ҽ����е��
�����������ע���Ӫҵ�ص����ڵ�EEA��Ա�������ܻ��� (Competent Authority) ����ע�ᣬ�����(����������һ)��
- ���� I�� ҽ����е�� ����ʽ (custom-made) ҽ����е���������Լ��Ĺ�˾������̱�����Ͷ����EEA�г���
- ��ȫ����I��ҽ����е����Ϊһ�������ֳɵ�(I��)ҽ����е������ǩ�����ڰ����������Լ��Ĺ�˾��(���̱�)Ͷ����EEA�г���
- ���г������еĴ���CE��־��ҽ����е����ԭ�����̵�Ԥ��ʹ��Ŀ�ĺ�ʹ�÷�Χ�ڣ������Լ��Ĺ�˾��(���̱�)����һ��ϵͳ��һ�������֮�У�
- Ϊ���������Լ��Ĺ�˾��(���̱�)Ͷ����EEA�г����������������������������ſ�ʹ�õ�ϵͳ�����������CE��־��ҽ����е��������������
- ��λ��ŷ������EEA�����ҽ����е�����̵�ŷ����Ȩ������
�������ҽ����е�����̵�����ŷ������EEA����û��ע���Ӫҵ��ַ�Ļ���������ָ����ŷ������EEA�Ļ�Ա������ע��Ӫҵ��ַ��һ����Ȩ�������������ְ��
����ʽ (custom-made) ҽ����е��ָ����һ��רҵ����д�Ĵ�����Ϊijһ���ض��IJ���ר��ʹ�÷�������������������е��
- �����������ҽ����е(IVD):
�����������ע���Ӫҵ�ص����ڵ�EEA��Ա�������ܻ��� (Competent Authority) ����ע�ᣬ�����(����������һ)��
- �����������ҽ����е(IVD)���������Լ��Ĺ�˾������̱�����Ͷ����EEA�г���
- �����Լ��Ĺ�˾������̱����������������õ��������ҽ����е(IVD)��
- ��һ����ŷ������EEA����û��ע��Ӫҵ��ַ���������ҽ����е(IVD)�����̵�ŷ����Ȩ������
�������ŷ������EEA����û��ע���Ӫҵ��ַ����ϣ�����������ҽ����е(IVD)Ͷ��EEA�г��Ļ���������ָ����ŷ������EEA�Ļ�Ա������ע��Ӫҵ��ַ��һ����Ȩ�������������ְ��
About CE Marking:
�������Ҫ�������CE��֤����Ϣ����ӭ�������ǵ���������������.
|