Wellkang is a registered EU Authorised Representative (EC Rep) in the European Commission's EU medical device database EUDAMED under a unique Single Registration Number (SRN): XI-AR-000001836 for the markets of EEA/EU27 & Northern Ireland.
Wellkang is also a registered UK Responsible Person (UKRP) in the UK MHRA medical device database for the market of Great Britain: England, Wales & Scotland, after 1 Jan 2021, which is no longer part of the EU single market after Brexit.

Are you ready for Brexit? Do you have a Brexit contingency plan?
You may need either an EU/EC European authorized representative based in EU-27 countries or a UK authorised representative (so-called "UK Responsible Person") based in UK, or may even need both EU & UK representatives, depending on different brexit scenarios.
Register/Notify your MD-Medical Devices & IVD-In Vitro Diagnostic Medical
Devices with MHRA in UK & other EEA (EU/EFTA) authorities by world-leading
consultancy- Wellkang team based in both UK (England) & EU-27 (Ireland).
Wellkang team can help you under all Brexit scenarios!
Click here to get FREE Guide Now! |
FAQ/Q&A: Questions and Answers about CE Marking of Medical Devices
Legal basis for medical devices
Directives
Medical Devices are regulated in EEA- European Economic Area (EEA=EU+EFTA, totally 30 member states) by 3 New Approach Directives.
- Active Implantable Medical Device Directive, AIMDD (90/383/EEC)
- (General) Medical Device Directive, MDD (93/42/EEC)
- In Vitro Diagnostic Medical Device Directive, IVDMDD (98/79/EC)
Authorities
Each EEA member state specifies some Competent Body(ies) (CB), also called "Competent Authority(ies)" to enact the directive within its territory. Each CB can specify one or more Notified Bodies (NB), to act as third party accessors of the manufacturer´s compliance. A NB may be Notified for accessing the products under all allowed conformity modules under the particular directive or only part of modules. Therefore, it is very important to select the most appropriate NB according to the manufacturer's own need.
Other Related Directives
Some other important directives may relate to the Medical Devices are:

Is my product a Medical Device (MD), an In Vitro Diagnostic Medical Device (IVD) or an Active Implantable Medical Device (AIMD)?
A Medical Device (MD)
is defined in Directive (93/42/EEC) as: Any instrument, apparatus, appliance, material or other article, whether used alone or in combination, including the software necessary for the proper application, intended by the manufacturer to be used for human beings for the purpose of :
- diagnosis, prevention, monitoring, treatment or alleviation of disease,
- diagnosis, monitoring, treatment, alleviation of or compensation for an injury or handicap,
- investigation, replacement or modification of the anatomy or of a physiological process,
- control of conception,
and which does not achieve its principal intended action in or on the human body by pharmacological, immunological or metabolic means, but which may be assisted by such means.
Attention:
According to Directive 2007/47/EC which, will become mandatory on 21 March 2010, has amended the Directive 93/42/EEC, Medical Device means: any instrument, apparatus, appliance, software, material or other article, whether used alone or in combination, including the software intended by its manufacturer to be used specifically for diagnostic and/or therapeutic purposes and necessary for its proper application, intended by the manufacturer to be used for human beings for the purpose of:
- diagnosis, prevention, monitoring, treatment or alleviation of disease,
- diagnosis, monitoring, treatment, alleviation of or compensation for an injury or handicap,
- investigation, replacement or modification of the anatomy or of a physiological process,
- control of conception,
and which does not achieve its principal intended action in or on the human body by pharmacological, immunological or metabolic means, but which may be assisted by such means.
|
For a number of products it is not clear if they are medical devices or not. There are a number of examples of products that may or may not be medical devices depending on the Intended Purpose for Use, assigned by the manufacturer to the products.
The following Toiletry and Cosmetics Products can also be Medical Devices if a medical claim is being made by the manufacturer for the device, although these products are usually not Medical Devices:
- tooth brushes, dental sticks, dental floss, dental chewing gums;
- baby nappies, hygiene tampons, mattress protectors;
- contact lenses intended to provide colour to the eyes;
- instruments for tattooing;
- deodorants for use with devices;
- wigs.
An In Vitro Diagnostic Medical Device (IVD)
is defined in Directive (98/79/EC) as: any medical device which is a reagent, reagent product, calibrator, control material, kit, instrument, apparatus, equipment, or system, whether used alone or in combination, intended by the manufacturer to be used in vitro for the examination of specimens, including blood and tissue donations, derived from the human body, solely or principally for the purpose of providing information:
- concerning a physiological or pathological state, or
- concerning a congenital abnormality, or
- to determine the safety and compatibility with potential recipients, or
- to monitor therapeutic measures.
Specimen receptacles are considered to be in vitro diagnostic medical devices. 'Specimen receptacles` are those devices, whether vacuum-type or not, specifically intended by their manufacturers for the primary containment and preservation of specimens derived from the human body for the purpose of in vitro diagnostic examination.
Products for general laboratory use are not in vitro diagnostic medical devices unless such products, in view of their characteristics, are specifically intended by their manufacturer to be used for in vitro diagnostic examination;
An Active Implantable Medical Device (AIMD)
is defined in AIMDD Directive (90/383/EEC) as:
"Any medical device which is intended to be totally or partially introduced, surgically or medically, into the human body or by medical intervention into a natural orifice, and which is intended to remain after the procedure".
According to the directive, a device is active if it relies for its functioning on a source of electrical energy or any source of power other than that directly generated by the human body or gravity. This includes, for instance, devices activated by means of pressure, unless this effect is achieved by energy resulting from the patients body. The definition implies that the function of the device involves using the source of power to perform useful work. Mere transmission of heat, light, pressure or vibration does not mean that a device is active.
Examples of types of devices normally covered by this directive include:
- implantable cardiac pacemakers
- implantable defibrillators
- leads, electrodes, adaptors for the above
- implantable nerve stimulators
- bladder stimulators
- sphincter stimulators
- diaphragm stimulators
- cochlear implants
- implantable active drug administration devices
- catheters, sensors for active drug administration devices
Which classification does a Medical Device (MD) fall into?
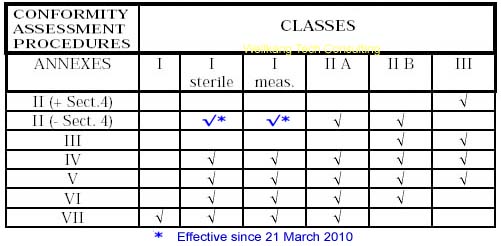
A medical device (MD) may be classified as Class I (including Is & Im), Class IIa, IIb and III, with Class III covering the highest risk products. The higher the classification the greater the level of assessment required. All Class Is, Im, IIa, IIb and III medical devices require the intervention of third party: the so-called Notified Body.
The classification rules are set out in Annex IX of the directive. This annex includes definitions of the terminology used in the classification rules. Classification of a medical device will depend up on a series of factors, including:
- how long the device is intended to be in continuous use
- whether or not the device is invasive or surgically invasive,
- whether the device is implantable or active
- whether or not the device contains a substance, which in its own right is considered to be a medicinal substance and has action ancillary to that of the device.
It is usually the Intended Purpose for Use that determines the class of the medical device and not the particular technical characteristics of the device.
It is the Intended Purpose for Use, assigned by the manufacturer to the device, that determines the class of the medical device and not the class assigned to other similar products manufactured by the same manufacturer or different manufacturers. For instance, two sutures that have the same composition may well have different Intended Purpose.
A manufacturer may successfully avoid the particular higher classification by clearly define on the labelling the Intended Purpose in such a way that the device falls into the lower class!
Read more: Class I (including Is & Im) medical devices CE Marking procedures
For more information about classification rules, please visit Guidelines for Classification of Medical Devices.
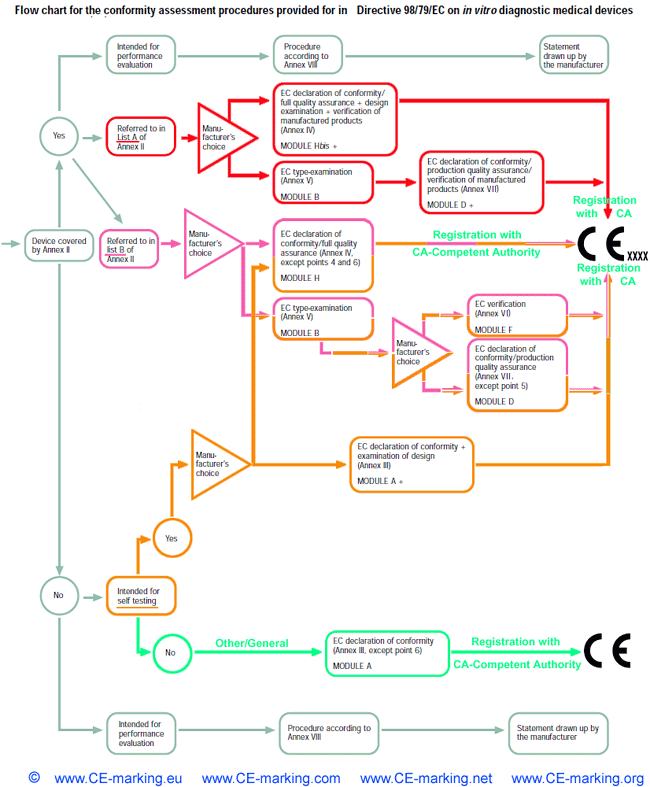
Which category does an In Vitro Diagnostic Medical Device (IVD) fall into?
The IVD Directive (98/79/EC) groups IVDs into four categories. These categories are, in
order of increasing perceived risk:
- Other/General Device:
all devices except Annex II and self-testing devices;
- Device for Self-Testing (not listed in Annex II):
a device intended by the manufacturer to be able to be used by lay persons in a home environment, excluding self-test devices
covered in Annex II;
- Device of List B, Annex II
of the Directive: which, amongst others, includes reagents and products for rubella, toxoplasmosis and phenylketonuria as well
as devices for self testing for blood sugar;
- Device of List A, Annex II
of the Directive: which includes reagents and products for human immunodeficiency virus I and II, hepatitis B, C and D.
Further info is available at
IVD-In Vitro Diagnostic medical devices CE Marking procedures
In Vitro Diagnostic Medical Device Directive, IVDMDD (98/79/EC).
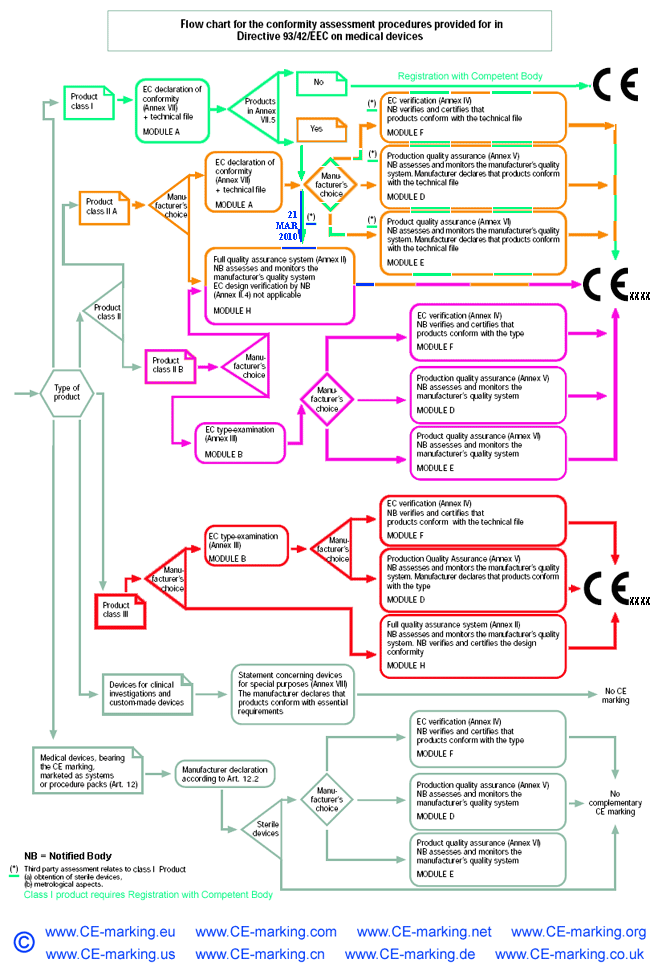
Medical device CE Marking procedures
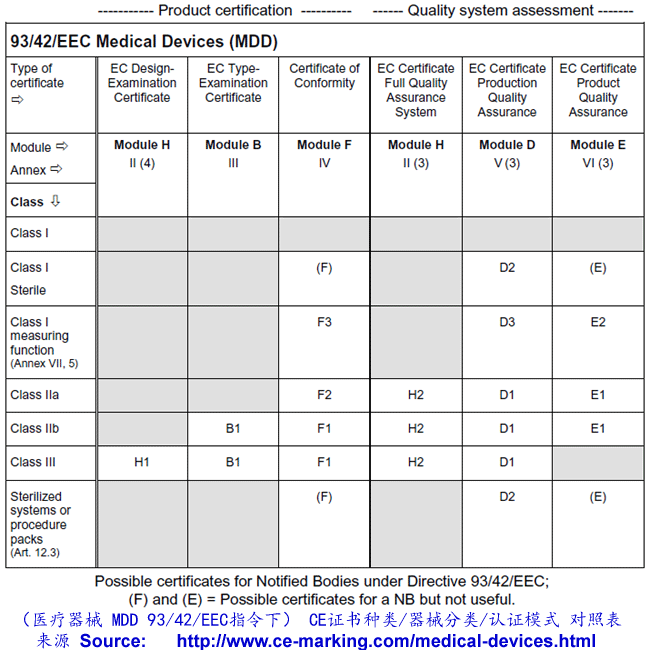
Depending on the individual case, each Medical Device manufacturer must choose its own and most suitable conformity module(s) which address(es) its particular product categories and business needs.
It is very important to select your European Authorized Representative like Wellkang before you choose a Notified Body from more than 100 of them based in 30+ countries; As your Auth Rep, Wellkang will particularly assist you in classifying your product before submission to the most suitable Notified Body.
Steps to obtain CE Marking for your Medical Devices
- Appoint Wellkang as your single European Authorized Representative within the 30 EU+EFTA countries.
- Wellkang assist you in identifying all EU Directives applicable to your product.
- Wellkang assist you in classifying your device.
- Wellkang assist you in selecting the most appropriate conformity assessment module.
- Wellkang assist you in selecting your Notified Body, if your chosen conformity module requires so, to perform the third-party conformity assessment tasks.
- Wellkang assist you in assessing your device according to Essential Requirements.
- Wellkang assist you in preparing the "Technical File".
- Wellkang assist you in preparing your "Declaration of Conformity".
- You affix the CE Marking on your device and start to sell.
- Wellkang EU Authorized Representative Service AFTER you have affixed CE Marking on medical device
Wellkang continue acting as your Auth Rep for vigilance and inspection purpose while your CE-Marked devices are placed on the European market by you directly or through your local European distributors. Our service mainly covers the follows:
- 10.1 Publish Your Device:
At webpage http://www.CEmark.info/mdd/YourProduct.html for third party verification
- 10.2 Product Registration:
If applicable, we can register your product in EU and get your product a Certificate of Registration.
- 10.3 Renewal and Update Product Registration:
The product Certificate of Registration is valid for one year only and must be renewed annually. The information about the product must be updated whenever it changes and at least once a year when renew the product Certificate of Registration.
- 10.4 Keep Your Technical Files:
We store and update the Technical Files of your products sold in Europe. The Technical Files may be inspected at any time by the Competent Authorities for a period ending at least five (5) years after the last product has been manufactured.
- 10.5 Legislation Monitoring:
We monitor and report on new developments in European product legislation relevant to your products.
- 10.6 Vigilance and Incident Reporting:
We assist with product vigilance and incident reporting.
- 10.7 Product Recalls and Advisory Notices:
We assist with Product Recalls and the issuing of Advisory Notices
Why must a non-EEA manufacturer of medical device appoint a European Authorized Representative?
The European Union's 'New Approach Directives' are mandatory on all member countries to enact through national legislation. This legislation requires manufacturers to display  Marking on their products, packaging and accompanying literature. Where a new approach directive is in force, it is (with few exceptions) "an offense to place a product on the market without Marking". The manufacturer is legally responsible for ensuring that the product confirms to the requirements of the directive and for applying Marking. Marking on their products, packaging and accompanying literature. Where a new approach directive is in force, it is (with few exceptions) "an offense to place a product on the market without Marking". The manufacturer is legally responsible for ensuring that the product confirms to the requirements of the directive and for applying Marking.
For medical devices, it is compulsory, under the 3 Medical Devices Directives, that the non-EEA manufacturer must appoint an Authorized Representative (authorised representative) which must be established (have a registered business) in the EEA (28 EU + 3 EFTA) member states to act on manufacturer's behalf in carrying out certain tasks including:
- Dealing with the regulatory authority;
- Keeping, for a period extending to at least five (5) years after manufacture of the product, the updated Technical Documentation (or Technical File) available, in a timely fashion when called upon, for review and inspection by EEA Competent and/or Surveillance Authorities;
- Register, Update, and/or Notify class I, and IVD, Medical Devices with the EEA Competent Authorities;
- Assisting in Vigilance/Incident Reporting, Product Recalls and the issuing of Advisory Notices.
To maintain the independence from distributors and importers, many non-EEA manufacturers prefer to use Wellkang, a world-leading CE marking consultancy, as the European Authorized Representative acting as the central point to the government authorities in all 30 EU+EFTA member states, therefore enjoy an effective and professional communication with the authorities and most favorable outcome for their businesses.
As of 21 March 2010, a Single EEA European Authorised/Authorized Representative must be designated by a non-EEA manufacturer of medical devices as required by Article 14.2* of Council Directive 93/42/EEC amended by Directive 2007/47/EC.
*Article 14.2 of Council Directive 93/42/EEC as amended by Directive 2007/47/EC:
Where a manufacturer who places a device on the market under his own name does not have a registered place of business in a Member State, he shall designate a single authorised representative in the European Economical Area (EEA). For devices referred to in the first subparagraph of paragraph 1, the authorised representative shall inform the competent authority of the EEA member State in which he has his registered place of business of the details referred to in paragraph 1.
Why must inform/notify/register with the Competent Authority?
Both the Medical Devices Directive (93/42/EEC) and In Vitro Diagnostic Medical Device Directive (98/79/EC) require manufacturers or, their authorised representatives or others placing medical device(s) on the EEA market, to provide certain information to the Competent Authorities in the EEA Member State where they have a registered place of business. These requirements have been transposed into national laws of the EEA Member States.
After the receipt of the notification/registration, the Competent Authorities shall process the data and inform the Commission of the European
Communities and the other States Party to the Agreement on the European Economic Area, upon request.
When to inform/notify/register with the Competent Authority?
- For General Medical Devices:
You must inform the Competent Authority when you first apply the CE marking to your
class I device(s). If you have a number of models and the CE marking procedure is
being introduced over a period, you may if you wish, inform the Competent
Authority of all devices when informing of the first CE marking.
Manufacturers of custom-made devices, systems or procedure packs, and
sterilisation companies, should register no later than the first time they claim
compliance with the Directive.
- For In Vitro Diagnostic Medical Devices (IVDs):
For In Vitro Diagnostic Medical Devices there is a transitional period until 7 December 2003 in respect of a device placed on the market and a transitional period until 7 December 2005 in respect of a device put into service, during which a manufacturer can choose either to follow existing national regulations to apply the CE mark. You must inform the Competent Authority when you first apply the CE marking to your IVDs.
Who should apply/register with the Competent Authority?
The EEA Competent Authorities usually charge fees for the registration/notification.
- For General Medical Devices:
You must register with the Competent Authority in an EEA Member State in which you have your registered place of business if you:
- manufacture class I or custom made devices and place them on the market under your own name, or trading name(s);
- fully refurbish class I devices, or label one or more ready made devices, with a view to placing them on the market under your own name;
- place devices bearing the CE marking on the market, under your own name in a system or a procedure pack within their intended purposes and within the limits of uses specified by their original manufacturers;
- sterilise, for the purpose of placing on the market under your own name, systems or procedure packs or other CE marked medical devices designed by the manufacturer to be sterilised before use;
- are the authorised representative of a manufacturer who does not have a registered place of business in the EEA.
If you do not have a registered place of business in the EEA Member State you must designate an authorised representative established in the EEA to act on your behalf.
Custom Made Devices mean devices manufactured in accordance with a professionals written prescription for the sole
use of a particular patient and are not adapted from mass produced items.
- For In Vitro Diagnostic Medical Devices (IVDs):
You must register with one Competent Authority in a Member State in which you have your registered place of business if you:
- manufacture in vitro diagnostic medical devices (IVDs) and place them on the market under your own name, or trading name(s);
- manufacture IVDs for performance evaluation and make them available under your own name, or trading name(s);
- are the authorised representative of a manufacturer who does not have a registered place of business in the Community.
If you do not have a registered place of business in a Member State and you wish to place IVDs on the European market then you must designate a person established in the Community to act on your behalf as your Authorised Representative.
Changes to registered details?
After the product registration or notification, the manufacturer or its authorised representative must inform the Competent Authority about any changes or additions to the registered details. Additionally, the Competent Authority will regularly review the records and request updating/confirmation of the registered information. The EEA Competent Authorities usually charge a fee for the product updating (change of registration).
Cost and fees
The cost and fees involved in the CE marking of Medical Devices will normally include the follows:
- Certification Fees charged by Notified Bodies
Notified Bodies always charge fees for issuing EC Certifications, the fees charged by the Notified Bodies, which are in general profit-driven organizations in the private sector, varies from Notified Body to Notified Body, from country to country.
- Official Fees charged by governmental Competent Authorities
The EEA Competent Authorities, which are in general non-profit governmental agencies in the public sector, normally charge fees for the processing and administration of the registration/notification of manufacturers and devices of MDs/IVDs. The procedures and official fees vary greatly from Member State to Member State among the 30 EEA countries. For instance,
- in UK,
the Competent Authority charges 100GBP (about US$135) each time for registration, notification, update or change of registered information;
- in Spain,
the Competent Authority charges a fee starting at 455.12Euro (about US$705) per product for registration/notification;
- in Sweden,
the Competent Authority charges annually a Registration Fee of 2150SEK (about US$320) per manufacturer, plus a Registration Fee, based on number of products, starting at 1000SEK (about US$150) for 1 to 10 products.
Click here NOW ! to learn more about the registration/notification procedures and official fees charged by the Competent Authorities in 30 EEA member states.
- Authorised Representative Fees
Non-EEA manufacturers MUST appoint an Authorised Representative who has a registered business in EEA countries. The Auth Rep fees may vary depending on the level of services, knowledge & reputation of the firm, taxation of the country domiciled, etc. As a world-leading consultancy in CE marking, Wellkang is proudly present you with high-standard of services at competitive prices. Click here to view our Auth Rep fee list.
* The European Economic Area (EEA):
The EEA includes EU countries and also Iceland, Liechtenstein and
Norway. It allows them to be part of the EEA single market.
Switzerland is a member of EFTA, but neither an EU nor EEA member. It however is still part of the single market.
About CE Marking:
| Home | About Us | Contact Us | Copyright & Disclaimer | Privacy | Log-in | Order Now | |
